This module rely on adding niches and spatial domains to AnnData metadata. It relies on two python packages:
Cell Charter
Varonne M. et al. CellCharter reveals spatial cell niches associated with tissue remodeling and cell plasticity. Nature Genetics volume 56, pages 74–84 (2024)
* Article
* Documentation
Novae
Blampey Q. et al. Novae: A Graph-Based Foundation Model for Spatial Transcriptomics Data. Mol Syst Biol. 2019;15(6):e8746. Published 2019 Jun 19. doi:10.15252/msb.20188746
* Article
* Documentation
01. Cell Charter niches analysis
The code below is based on the following CellCharter tutorial notebook.
import cellcharter as cc
import scispy as scis
import torch
import scvi
device = torch.device('cuda:0' if torch.cuda.is_available() else 'cpu')
print('torch version is {}, device is {}'.format(torch.__version__, device))
print('scanpy version is {}, anndata version is {}'.format(sc.__version__, ad.__version__))
print('squidpy version is {}'.format(sq.__version__))
print('scvi version is {}'.format(scvi.__version__))
print('cellcharter version is {}'.format(cc.__version__))
adata = ad.read_h5ad('000-outs/ad6_scanvi_scmusk.h5ad')
adata.X = adata.layers["counts"].copy()
sc.pp.normalize_total(adata)
sc.pp.log1p(adata)
scvi.settings.seed = 12345
scvi.model.SCVI.setup_anndata(
adata,
layer="counts",
batch_key='sample'
)
model = scvi.model.SCVI(adata)
model.train(early_stopping=True, enable_progress_bar=True)
adata.obsm['X_scVI'] = model.get_latent_representation(adata).astype(np.float32)
sq.gr.spatial_neighbors(adata, library_key='sample', coord_type='generic', delaunay=True, spatial_key='spatial')
cc.gr.remove_long_links(adata)
cc.gr.aggregate_neighbors(adata, n_layers=3, use_rep='X_scVI', out_key='X_cellcharter', sample_key='sample')
gmm = cc.tl.ClusterAutoK(
n_clusters=(10,30),
max_runs=10,
model_params=dict(random_state=12345, trainer_params=dict(accelerator='gpu', devices=1))
)
gmm.fit(adata, use_rep='X_cellcharter')
adata.obs['cc_niches'] = gmm.predict(adata, use_rep='X_cellcharter')
adata.write('000-outs/ad6_scanvi_scmusk_cc.h5ad')
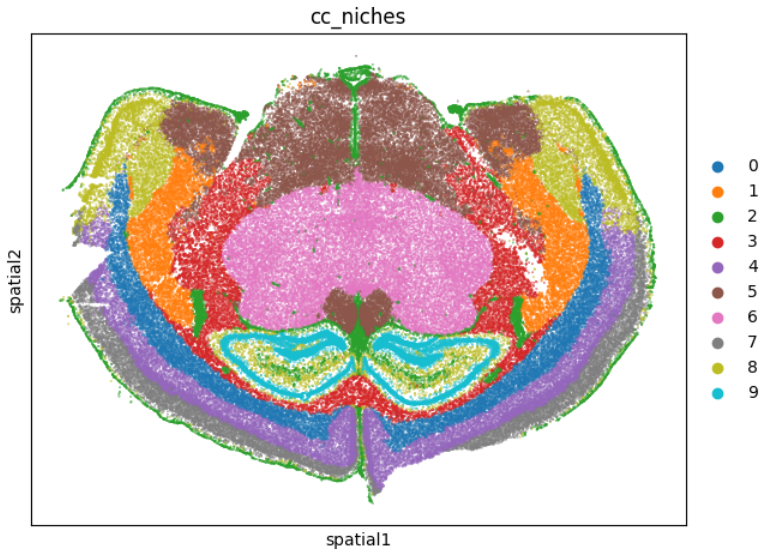
sub = adata[adata.obs['sample'] == '2658']
sq.pl.spatial_scatter(sub, color="cc_niches", shape=None, size=0.1)
02. Novae spatial domains analysis
import anndata as ad
import scanpy as sc
import novae
adata = ad.read_h5ad('000-outs/ad6_scanvi_scmusk_cc.h5ad')
novae.utils.spatial_neighbors(adata, slide_key='sample', radius=80)
novae.plot.connectivities(adata)
# select foundation model to use, here mouse brain
model = novae.Novae.from_pretrained("MICS-Lab/novae-brain-0")
# Option 1: zero-shot
model.compute_representations(adata, zero_shot=True)
# Option 2: fine-tuning
#model.fine_tune(adata, accelerator='cuda')
#model.save_pretrained(save_directory="./my-model-directory")
model.compute_representations(adata, accelerator='cuda')
# 7 spatial domains by default, here asking for 15
model.assign_domains(adata, level=15)
novae.plot.domains(adata)
adata.write('000-outs/ad6_scanvi_scmusk_cc_novae.h5ad')
# novae.plot.pathway_scores(adata[adata.obs['sample'] == 'C31'], pathways="h.all.v2024.1.Hs.json", figsize=(10, 7))
# novae.plot.pathway_scores(
# adata, pathways="h.all.v2024.1.Hs.json", figsize=(4, 4), slide_name_key="sample", pathway_name='HALLMARK_COMPLEMENT',
# )